mobility-model-minimos
The mobility model used in
MINIMOS 6 is used to simulate the doping dependent
mobility in Si and takes into account the scattering of the carriers by charged
impurity ions which leads to a degradation of the carrier mobility (ionized
impurity scattering). This model
is temperature dependent and takes into account the reduced mobility due to
lattice scattering (i.e. the values in the database under keyword $mobility-model-constant are the same as under this keyword).
It is a model that combines lattice and impurity scattering.
The formula of Caughey and Thomas (D. Caughey, R. Thomas,
Carrier Mobilities in Silicon Empirically Related to Doping and Field,
Proc. IEEE 55, 2192 (1967))
is used together with temperature dependent coefficients. This model is well
suited for Si.
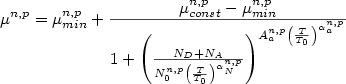
The total concentration of ionized impurities is given by ND+NA.
Note: In nextnano³ we use the nominal dopant
concentration as specified in the input file and not the ionized one.
For T >= 200 K:
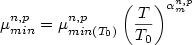 (1)
(1)
For T < 200 K:
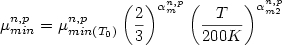 (2) (2)
By setting
alphamn
= alpham2n,
alphamp
= alpham2p
and
alphaan
= alphaap
= 0
equation (2) reduces to equation (1) and this model can also
be applied to other basic materials.
The parameter values used in this model for electrons and holes,
respectively, are taken from the PhD thesis of V. Palankovski "Simulation
of Heterojunction Bipolar Transistors" (TU Vienna). (Note: The
exponents n-gamma-lattice-temp, p-gamma-lattice-temp
have opposite sign in his PhD thesis.)
| Parameter in
formula |
Description |
Input parameter |
| electrons |
holes |
|
electrons |
holes |
| µminn(T0) |
µminp(T0) |
reference mobility parameter |
n-mu-min |
p-mu-min |
| alphamn |
alphamp |
exponent |
n-alpha-mu-min |
p-alpha-mu-min |
| alpham2n |
alpham2p |
exponent |
n-alpha-mu-min2 |
p-alpha-mu-min2 |
| N0n |
N0p |
reference impurity parameter |
n-n0 |
p-n0 |
| alphaNn |
alphaNp |
exponent |
n-alpha-n0 |
p-alpha-n0 |
| Aan |
Aap |
reference exponent parameter |
n-a-a |
p-a-a |
| alphaan |
alphaap |
exponent |
n-alpha-a-a |
p-alpha-a-a |
| µconstn |
µconstp |
result of $mobility-model-constant |
|
|
| ND |
NA |
concentration of ionized donors/acceptors |
|
|
| T |
T |
lattice temperature |
|
|
| T0 |
T0 |
temperature (300 K) |
|
|
!-------------------------------------------------------------!
$mobility-model-minimos
optional !
!
material-name
character
required !
number-of-parameters
integer
required !
!
!++++++++++++++++++++++++++++++++++++
! for binaries, e.g. GaAs
!++++++++++++++++++++++++++++++++++++
n-mu-lattice-temp
double
optional ! [cm2/Vs] same as
$mobility-model-constant
n-gamma-lattice-temp
double
optional ! []
same as
$mobility-model-constant
(but different sign as in MINIMOS documentation)
n-mu-min
double
optional ! [cm2/Vs]
n-alpha-mu-min
double
optional ! []
n-alpha-mu-min2
double
optional ! []
n-n0
double
optional ! [cm-3]
n-alpha-n0
double
optional ! []
n-a-a
double
optional ! []
n-alpha-a-a
double
optional ! []
!
p-mu-lattice-temp
double
optional ! [cm2/Vs]
$mobility-model-constant
p-gamma-lattice-temp
double
optional ! []
same as
$mobility-model-constant
(but different sign as in MINIMOS documentation)
p-mu-min
double
optional ! [cm2/Vs]
p-alpha-mu-min
double
optional ! []
p-alpha-mu-min2
double
optional ! []
p-n0
double
optional ! [cm-3]
p-alpha-n0
double
optional ! []
p-a-a
double
optional ! []
p-alpha-a-a
double
optional ! []
!
!
!++++++++++++++++++++++++++++++++++++
! for ternaries, e.g. Al(x)Ga(1-x)As
!++++++++++++++++++++++++++++++++++++
n-bow-mu-lattice-temp
double
optional ! [cm2/Vs]
n-bow-gamma-lattice-temp
double
optional ! []
n-bow-mu-min
double
optional ! [cm2/Vs]
n-bow-alpha-mu-min
double
optional ! []
n-bow-alpha-mu-min2
double
optional ! []
n-bow-n0
double
optional ! [cm-3]
n-bow-alpha-n0
double
optional ! []
n-bow-a-a
double
optional ! []
n-bow-alpha-a-a
double
optional ! []
!
p-bow-mu-lattice-temp
double
optional ! [cm2/Vs]
p-bow-gamma-lattice-temp
double
optional ! []
p-bow-mu-min
double
optional ! [cm2/Vs]
p-bow-alpha-mu-min
double
optional ! []
p-bow-alpha-mu-min2
double
optional ! []
p-bow-n0
double
optional ! [cm-3]
p-bow-alpha-n0
double
optional ! []
p-bow-a-a
double
optional ! []
p-bow-alpha-a-a
double
optional ! []
!
$end_mobility-model-minimos
optional !
!-------------------------------------------------------------!
Syntax
material-name = Si
Name of material to which this set of parameters
applies. Name has to be listed in
$default-materials.
number-of-parameters = 18
Control parameter if the number of parameters provided
is the same as demanded.
The parameters are specified as shown in the tables above. There are two
sets, one for electrons (n-) and one for holes (p-).
!++++++++++++++++++++++++++++++++++++
! for ternaries, e.g. Al(x)Ga(1-x)As
!++++++++++++++++++++++++++++++++++++
The specifiers containing "*-bow-*" are for alloys, i.e.
ternaries.
By default, linear interpolation is used if the bowing parameter is zero.
material-name
= Al(x)Ga(1-x)As
number-of-parameters = 4
n-bow-mu-lattice-temp
= 0d0 ! [cm2/Vs]
Bowing parameters are zero if linear interpolation is used.
...
|